
Beyond the curve: Dr. Peter Lurie's COVID-19 blog
It all seemed to start innocently enough. But by the end of the week, the Food and Drug Administration’s reputation for science-based decision-making was in tatters, described by Harvard FDA historian Daniel Carpenter as “a low moment for the FDA in at least a generation.” How did that come to pass?
On Wednesday of the week before last, a seemingly obscure announcement appeared on the website of the Department of Health and Human Services (HHS), FDA’s parent agency. Titled, in inimitable bureaucratese, “Rescission of Guidances and Other Informal Issuances Concerning Premarket Review of Laboratory Developed Tests,” and clocking in at a scant 252 words, it seemed like the kind of innocuous pronouncement that matters to the cognoscenti but that would soon fade into obscurity.
But, in fact, it represented the end game in a war waged for years by the laboratory industry seeking to preclude FDA regulation of a major subset of laboratory tests, including those that could be useful in stemming the COVID-19 pandemic. The tests, called laboratory developed tests (LDTs), involve samples sent off to labs typically affiliated with academic centers that conduct the test on-site. Once a niche industry, it is now a major earnings center for many academic institutions.
Back in 2014, when I was at FDA, we used a document called a guidance to assert FDA jurisdiction over these tests, putting forth a risk-based approach to regulating this burgeoning industry. I even helped put together a set of 20 case studies where inaccurate LDTs may have caused actual harm to patients. That report, apparently missing from the agency’s website, is still available here.
Wednesday’s announcement made a seemingly technical change: henceforth all regulation of LDTs would have to occur through the painstaking process of device-by-device notice and comment regulation (not guidance), a clear attempt to hobble the agency’s oversight of LDTs, even in the context of a pandemic. But the new policy extends far beyond COVID-19 to encompass a wide arrange of diseases, from cancer to cardiovascular disease. The new policy assures that these tests will only be regulated under the much weaker authority available to the Center for Medicare & Medicaid Services, which in effect regulates labs, not tests.
Significantly, the political nature of the decision was reflected in its appearance only on the HHS website, evidently because FDA Commissioner Stephen Hahn had opposed the policy change.
That Wednesday also brought an extremely concerning White House press briefing statement from President Trump, one that proved to be a harbinger of things to come. Without evidence, a long-forsaken requirement for Presidential claims, the President explicitly injected politics into FDA’s review of convalescent plasma: “You have lot of people over there that don’t want to rush things. They want to do it after November 3rd,” he said in a White House press briefing.
Trump renewed the baseless charge via Twitter on the Saturday: “The deep state, or whoever, over at the FDA is making it very difficult for drug companies to get people in order to test the vaccines and therapeutics. Obviously, they are hoping to delay the answer until after November 3rd.” And, not to be outdone, White House Chief of Staff Mark Meadows weighed in on a Sunday morning kibbitz show saying FDA employees “need to feel the heat.”
Although Trump characterized the use of convalescent plasma as having an “incredible rate of success,” the data in question are observational only.
Which brings us to the Press Conference from Hell itself. Trump mounted the White House podium on the evening of the Republican convention to announce a “very historic breakthrough” related to the “China virus” and convalescent plasma, which is the antibody-rich material that can be isolated from the blood of patients recovering from COVID-19 and then infused into acutely ill patients. In fact, there was not much “historic” about the announcement. Although Trump characterized the use of convalescent plasma as having an “incredible rate of success,” the data in question were drawn from observations of treated patients – not patients randomized to receive or not receive convalescent plasma as is typically required to make such claims. This means that there was no untreated group to which to compare the therapy and that factors other than the treatment could explain any differences observed. As readers of this blog well know, only randomized, controlled trials can provide sufficient evidence to conclusively prove a product’s effectiveness.
But this is where FDA Commissioner Stephen Hahn picked up the slack. After the requisite sycophantic thanking of the President for his leadership, he went on to describe the study results. In fact, the information presented was so fragmentary that observers are still scratching their heads trying to figure out where his assertions came from, as they didn’t appear in any documents FDA provided at the time.
But here’s our best guess. The major study on convalescent plasma was put together by the Mayo Clinic, which has data from some 100,000 treated patients. The findings from 35,322 of those were reported in a non-peer reviewed preprint available here. Because there was no untreated control group, the researchers compared patients who received convalescent plasma with high amounts of antibodies to those who received plasma with low amounts and found that the 7-day mortality rates were 8.9% and 13.7%, respectively. (These differences were more impressive than the 30-day mortality data, which the administration chose not to emphasize.)
Time for this blog to go full-on nerd again. Broadly, there are two ways to analyze those numbers. One would be to divide one into the other (8.9/13.7) which is 0.65 or 65%, or a 35% reduction in mortality from the 13.7% in the low-antibody group. This is called a relative risk reduction. The alternative would be to subtract the two numbers (13.7-8.9) yielding an absolute risk reduction of 4.8%. Both of these numbers can be appropriate ways of depicting the findings, though it’s well known that clinicians find relative risk reduction presentations more convincing than absolute risk presentations based on the same data.
Now let’s go to the pictures. In the figures below, we’ve rounded the numbers to be whole numbers. We start with a population of 100 patients who received convalescent plasma. According to the Mayo data (and disregarding for now the absence of a control group or randomization), 9% (formerly 8.9%) of patients will die regardless of whether they receive high- or low-antibody plasma (in yellow), 5% (the absolute reduction) will survive because they received high- instead of low-antibody plasma (in green), and the rest will survive regardless of the treatment they receive (in red). It looks like this, with each square representing one of the 100 hypothetical patients:
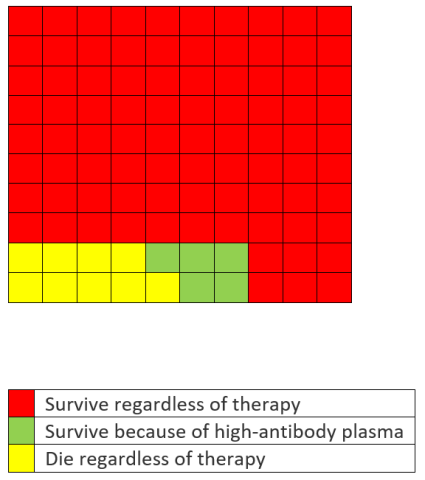
But Hahn said something different. After establishing his credentials as a cancer specialist, he stated that, “What that means is … 100 people who are sick with COVID-19, 35 would have been saved because of the administration of plasma.” But, as the graphic above shows, the actual number that would have been saved is 5, not 35. In other words, he used the 35% relative risk reduction and expressed it as an absolute risk reduction.
Here’s what his data would have looked like graphically, assuming the same 8.9% mortality rate in the low-antibody group (with 35 green squares).
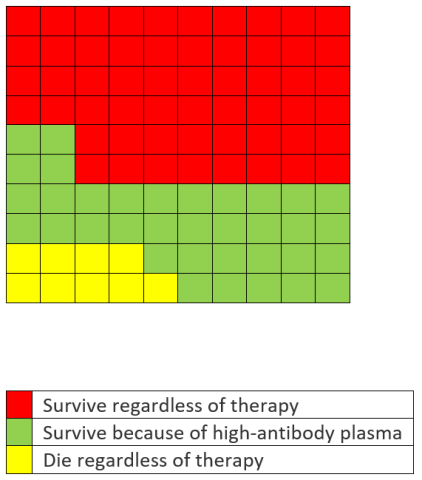
Oops. Even Trump characterized the 35% reduction more accurately than the cancer specialist. Under pressure from a number of former FDA Commissioners (and us), Hahn admitted 24 hours later that he had erred. But his performance was an embarrassment as he left the President’s own exaggerations uncorrected and failed to defend the agency when Trump asserted that there are people at the FDA “that can see things being held up ... and that’s for political reasons.”
This being Trumpland, heads rolled later in the week. Wayne Pines, a consultant who had proffered the morally sound advice to apologize for one’s mistakes, was soon let go, having violated a core precept of the Trump administration to never apologize for anything.
A more deserving victim was Emily Miller, who only 11 days earlier had been appointed as Assistant Commissioner for Media Affairs (she said that God was directing her path to this position), a post not typically held by political appointees. Armed with no scientific or medical bona fides, she was better known for a gun rights book called “Emily Gets Her Gun: But Obama Wants to Take Yours,” based in part on a robbery she experienced, although Erik Wemple at the Washington Post has pointed out the shifting details of her account. She apparently had a role in FDA’s trumpeting of its actions in starkly partisan terms, a dramatic departure from the scientific (dare I say boring?) tone that typically characterizes FDA press releases: “Another Achievement in Administration’s Fight Against Pandemic.” She also defended Hahn’s misstatements. For this she was ousted, consigning her to Anthony Scaramucci territory for short-lived tenures in the current administration.
In any event, the FDA action was no more than a modest administrative maneuver, not a product approval. The product, which had previously only been available under something called an Expanded Access Protocol (don’t worry, expanded access has a recurring role in this drama and we’ll explain it more fully below) would now be available under an Emergency Use Authorization (EUA), the very mechanism abused by FDA in its handling of hydroxychloroquine. Perhaps this latest Trump Administration endorsement will promote access to the product, although about 100,000 hospitalized patients had managed to receive it under expanded access anyway. But it’s unlikely to hasten the completion of much-needed randomized controlled trials as patients are likely to elect to receive the product rather than take a 50-50 chance of getting it in a randomized, controlled trial.
Moreover, the EUA was granted over the objections of senior NIH officials, including Anthony Fauci, and despite the opinion of FDA’s own member of the administration’s vaccine and therapeutics initiative Operation Warp Speed, Janet Woodcock, who said that convalescent plasma had not been “proven as an effective treatment.”
In any event, the FDA action was no more than a modest administrative maneuver, not a product approval.
By this point in the week, the Republican convention was in full fact-free fervor. And FDA came to play an unwitting role in that, too. Assuming the podium in Kimberly Guilfoyle red, a woman named Natalie Harp, a member of the Trump campaign’s advisory board, literally asserted that the President had saved her life: “They didn’t give me the right to try experimental treatments, Mr. President. You did, and without you, I’d have died waiting for them to be approved.”
She was referring to her efforts to secure an investigational therapy for her Stage 2 bone cancer; Right to Try is a law signed by the President in May 2018 that purportedly eased access to such therapies for patients like Harp. In fact, FDA has long had a process for providing access to such therapies called Expanded Access, which grants some 99% of applications and does so in hours or days, as I testified before Congress when I was still at FDA.
And in an echo of Emily Miller, Natalie Harp’s story had its own dubious encounters with the truth. For one, Right to Try assured access only to drugs not approved by FDA; the one Harp obtained was approved, although not for her condition, an “off-label use” not covered by Right to Try, but covered by FDA’s Expanded Access Program. (By the way, it is estimated that no more than 10 patients have ever received a treatment through Right to Try though Trump had promised it would save “tremendous numbers of lives” when he signed it into law.) For another, she received the treatment at least two months before the bill was signed into law. Now that’s what we call a miracle cure! Needless to say, FDA did not step forward to correct the record.
With FDA thoroughly defiled, it was time to move on to CDC . Months ago, the agency released guidance that noted that “it is important that contacts of individuals with SARS-CoV-2 infection be quickly identified and tested.” This policy is critical as it forms the basis for the contact tracing so critical to stemming the pandemic – and it is estimated that 40% of infections are asymptomatic.
But the President has long chafed under his amply debunked belief that enhanced testing for the virus – rather than underlying increases in disease rates– fully explains the rise in COVID-19 cases over the summer. And the long lines for testing that persist are constant reminders that the President has failed to deliver on his promises of adequate testing. Turns out Trump alone can fix that: the President has advocated specifically to “slow the testing down, please.”
So when CDC changed its tune on asymptomatic testing, reportedly as a result of pressure “coming from the top down,” it came as no surprise. The revised CDC guidance suggests that such patients “don’t necessarily need a test” and referred journalists to HHS (see LDTs, above). CDC Director Robert Redfield (see our take on his scientific credentials here), never a stranger to awkward public pronouncements, tried to sooth frayed nerves in the public health community by saying that testing in that circumstance, “may be considered,” which helped, you know, not at all. The administration tried to insinuate that Anthony Fauci had endorsed the new guidelines but, referring to the meeting where the guidelines were finalized, he said, “I was under general anesthesia in the operating room and was not part of any discussion or deliberation regarding the new testing recommendations.” So much for that.
Late in the week, there was finally some good news. FDA announced an EUA for a test to detect viral protein that will reportedly cost only $5 and take 15 minutes to deliver results. For now, the EUA applies only to symptomatic patients, but, if the product proves effective in asymptomatic testing, it has major implications for contact tracing and the reopening of schools and workplaces. The FDA says the test has 97.1% sensitivity and 98.5% specificity, which are quite good, especially for a rapid test. (For an explanation of these terms and their implications, see our earlier blog.) The federal government, which elsewhere has asserted that testing is a state problem, snapped up 150 million of the promising tests.
Hopefully, this will put to bed suggestions that FDA lower its standards in order to increase the supply of tests. A key issue with relaxing standards is that doing so removes the incentives for manufacturers to make high quality tests – as opposed to those that can clear some reduced FDA approval bar. And, as I explained to WebMD here, low specificity, in particular, can create a major problem with false-positives – those who believe based on testing that they are positive but are not.
Hopefully, this will put to bed suggestions that FDA lower its standards in order to increase the supply of tests.
Do I have to keep repeating myself? The path out of this pandemic is not through crisis-justified deregulation, obsequious press releases, undergraduate-level mathematical errors, fact-challenged convention speeches or cynical revisions of guidances to “solve” problems of the administration’s own creation. We will emerge from this crisis (and emerge we will) when the public and institutions like prisons, nursing homes, and meatpacking plants (do I need to mention political gatherings?) comply with clear public health directions to socially distance, wear masks, quarantine when appropriate, contact trace and wash hands and when appropriately regulated companies produce the quality diagnostic, therapeutic and preventive products we so desperately need.
Contact Info: Contact us at cspinews[at]cspinet.org with questions, ideas, or suggested topics.